
ENFERMEDADES
Mieloma Múltiple
¿Qué es el Mieloma Múltiple?
El Mieloma Múltiple se caracteriza por la proliferación de células plasmáticas anormales en el interior de la médula ósea, que es la fábrica de las células de la sangre y que se encuentra en el interior de los huesos de la columna vertebral, el cráneo, la pelvis, la caja torácica y las áreas alrededor de los hombros y las caderas. Todos los sujetos normales tenemos células plasmáticas, en proporción no superior al 5%, las cuales se encargan de producir inmunoglobulinas que nos defienden de ataques externos por parte de virus, bacterias,…
En los pacientes con Mieloma, las células plasmáticas se convierten en anormales y además, proliferan de una manera incontrolada estando comprometidas a producir un único tipo de inmunoglobulina que es lo que llamamos componente monoclonal, pico monoclonal o para-proteína y que, según el tipo de proteína, hablamos de mieloma múltiple IgG, IgA, IgM, IgD o IgE, bien kappa o lambda en función del tipo de cadena pesada y ligera de la inmunoglobulina que producen las células plasmáticas.
Es la segunda enfermedad hematológica en orden de frecuencia tras los linfomas no Hodgkin y aparecen entre 4 y 5 casos nuevos por cada 100.000 habitantes por año aproximadamente. Afecta fundamentalmente a gente de edad avanzada y la edad media de aparición del mieloma está en torno a los 69 años. Sólo el 5-10% de los pacientes tienen menos de 40 años. Es más común en hombres y en algunos grupos raciales, como los afroamericanos.
El Mieloma Múltiple puede detectarse en un estadio precoz: las células plasmáticas anormales pueden proliferar en la médula ósea pero en una cantidad muy pequeña y de una manera muy lenta. El estadio más precoz se denomina Gammapatía Monoclonal de Significado Incierto (GMSI), en la que las células del mieloma constituyen menos del 10% de las células de la médula ósea y el componente monoclonal en el suero es inferior a 3 gramos por decilitro. El riesgo que la GMSI se convierta en Mieloma es muy bajo y además constante, 1% por año. Hay pacientes con una infiltración en la médula ósea por mayor número de células plasmáticas, entre 10% y hasta 60%, pero que también tienen un ritmo de proliferación lento y representan lo que se llama Mieloma indolente, asintomático o smoldering. El Mieloma indolente no es equivalente a la GMSI y es muy importante evaluar el riesgo de progresar a Mieloma porque hay pacientes con MM indolente de bajo riesgo, pero otros de riesgo intermedio que requieren más controles.
¿Qué lo causa?
No existe ninguna causa determinante de la aparición del Mieloma, aunque puede haber varios factores que pueden contribuir a que las células plasmáticas normales se transformen en malignas, como exposición a sustancias químicas tóxicas, la radiación atómica, situaciones que produzcan un descenso de la vigilancia inmune, o incluso exposición a algunos virus.
Hay cierta predisposición familiar para el mieloma múltiple: entre el 5% y 7% aproximadamente de los diagnósticos de mieloma se dan en un miembro de la familia que tiene un pariente cercano al que se le ha diagnosticado previamente de alguna enfermedad de las células plasmáticas, aunque no siempre sea un mieloma.
¿Cuáles son los signos y síntomas?
Como se ha mencionado anteriormente, las células plasmáticas normales producen inmunoglobulinas, las proteínas que nos defienden de ataques externos. Las células del mieloma no producen inmunoglobulinas normales por lo que se produce una alteración en el sistema inmunitario que se traduce en una mayor tendencia a desarrollar infecciones.
Muchos de los problemas o síntomas relacionados con el mieloma están derivados de la acumulación de células plasmáticas que proliferan en la médula ósea. Estos síntomas constituyen lo que clasicamente se conoce como CRAB y consiste en:
1) HiperCalcemia, es decir, elevación de los niveles de calcio en la sangre como consecuencia de la liberación de calcio al producirse destrucción ósea, causando confusión mental, deshidratación, estreñimiento, fatiga, debilidad.
2) Problemas Renales, ya que la proteína monoclonal producida por las células plasmáticas del mieloma se liberan al torrente sanguíneo y pueden pasar a la orina y dañar los riñones. Además, la elevación del calcio, así como la mayor tendencia a infecciones y otros factores pueden contribuir al desarrollo de insuficiencia renal, que se traduce en fatiga, confusión mental, disminución de la micción.
3) Anemia, que supone un descenso en el número y actividad de las células de la médula ósea encargada de producir hemoglobina. La anemia se manifiesta como fatiga, debilidad,.. y está también presente en casi el 80% de todos los pacientes con Mieloma.
4) Lesiones óseas (de Bone, hueso en inglés) como consecuencia de la activación por parte de las células plasmáticas a células que destruyen el hueso bloqueando a las células productoras del mismo. Estas lesiones óseas, que aparecen como “agujeros” en los huesos, o aplastamiento/fracturas vertebrales ocasionan dolor óseo, con inflamación, que puede condicionar incluso lesión neurológica si el aplastamiento o fractura vertebral afecta a la médula espinal. Está presente en casi el 80% de los pacientes con Mieloma y es el motivo más frecuente de consulta.
Se pueden producir otras manifestaciones clínicas como por ejemplo neuropatía (alteración en el funcionamiento de algunos nervios) si la proteína monoclonal se deposita en los nervios, infecciones recurrentes, hemorragias (si el componente monoclonal inhibe alguna de las proteínas que participan en la coagulación de la sangre).
También se producen síntomas como consecuencia de la proliferación local en la médula ósea: se produce una reducción en la producción de células sanguíneas y daños en el hueso circundante. Los resultados son las múltiples características comunes del mieloma antes descritas como anemia, predisposición a infecciones, dolor y fracturas óseas e hipercalcemia.
En relación con los síntomas del Mieloma, los pacientes deben conocer que recientemente se han incluído dentro de la definición o diagnóstico de Mieloma a un grupo de pacientes que no presentan ninguna de la sintomatología que antes se ha descrito, pero se sabe que poseen ciertas características (biomarcadores) que les identifica como pacientes con un riesgo inminente de presentar alguna de la sintomatología antes descrita. A estos pacientes se les considera ahora Mielomas porque pueden obtener un beneficio importante si reciben tratamiento. Los biomarcadores para identificar a estos pacientes son: infiltración en la médula ósea por al menos un 60% de células plasmáticas, la presencia de una relación entre las cadenas ligeras libres en suero clonales y las normales producidas por las células plasmáticas de la médula ósea superior a 100 o que aparezcan dos o más lesiones focales en la resonancia magnética nuclear (de 5mm o más de tamaño).
¿Cómo se diagnostica y se estadía?
El Mieloma se diagnostica detectando la infiltración por células plasmáticas anormales en la médula ósea de un paciente que, además, suele siempre presentar un componente monoclonal o paraproteína en el suero y/o orina junto con alguna de la sintomatología que antes se ha descrito, o si el paciente estuviera asintomático, por la presencia de alguno de los biomarcadores que lo identifican como un paciente con un riesgo inminente de presentar alguna sintomatología.
La mayoría de los pacientes con Mieloma presentan algún síntoma y el dolor es la manifestación más frecuente junto con la anemia que se traduce en fatiga, cansancio,… lo que condiciona la realización de un análisis de sangre inicialmente donde se puede observar un aumento de las proteínas totales en el suero del paciente. Ante esta elevación, se realiza un trazado electroforético para identificar qué proteína está elevada y se suele identificar lo que antes hemos definido como proteína monoclonal, o pico monoclonal. Ante este hallazgo y sabiendo que esta inmunoglobulina monoclonal está producida por células plasmáticas que están en la médula ósea, la siguiente exploración es un aspirado y/o biopsia de la médula ósea para identificar a las células responsables.
También, hay que realizar análisis adicionales para ver si el paciente tiene anemia, insuficiencia renal, hipercalcemia o lesiones óseas. Para detectar las lesiones óseas se realizan radiografías de todos los huesos del cuerpo, aunque recientemente esta técnica se está reemplazando por otras más sensibles como son la resonancia magnética nuclear, la tomografía axial computerizada (TAC) o el PET-TAC.
Con estas exploraciones se realiza el diagnóstico de Mieloma Múltiple añadiendo siempre el tipo en función de la inmunoglobulina monoclonal, IgG, IgA, IgM, IgD o IgE, y kappa o lambda.. La mayoría de los mielomas son IgG. Hay algunos pacientes en que las células plasmáticas producen sólo cadena ligera kappa o lambda y se elimina por la orina.
Cuando se diagnostica el Mieloma, la cantidad de enfermedad varía de un paciente a otro y su estimación es lo que se denomina estadiaje del mieloma. Clásicamente se utilizaba el sistema de Durie-Salmon que evalúa la relación entre la masa de mieloma y el daño causado, como enfermedad ósea o anemia. Sin embargo, el que más se ha venido utilizando es el sistema de estadíaje internacional (ISS, de International Staging System), resultado de la colaboración de más de 20 instituciones de investigación de todo el mundo. Este sistema está basado en la determinación de los valores de dos proteínas, la albúmina y la beta2 microglobulina. Los pacientes con una beta2 microglobulina elevada (superior a 5.5 mg/L) tiene un ISS de 3 e indica que podemos estar ante un mieloma más agresivo que aquellos pacientes que tienen niveles de albúmina (superior a 3.5 g/dL) y beta2 microglobulina normales (inferior a 3.5 mg/L) cuyo ISS sería de 1. En el medio estarían los pacientes con ISS 2, cuando no cumplen criterios de ISS 1 ni 3, cuyo pronóstico sería intermedio.
Además del ISS, se conocen otros factores pronósticos en el Mieloma que habitualmente se evalúan en los pacientes en el momento del diagnóstico, como son los niveles de lactato deshidrogenasa (LDH), o la citogenética/FISH (Hibridación in situ fluorescente) que consiste básicamente en evaluar los cromosomas de las células plasmáticas del paciente con Mieloma. Para ello se utilizan unos marcadores que asignan colores diferentes a cada cromosoma. Por ejemplo, si el cromosoma 4 está incorrectamente unido al cromosoma 14, entonces los dos puntos “coloreados” se ven juntos, lo que indica la presencia de una traslocación (4;14). También se pueden identificar pérdidas de un cromosoma entero o parte de él. En general, la presencia de anomalías cromosómicas no suele ser bueno desde el punto de vista pronóstico.
Recientemente, el sistema de estadíaje ISS antes mencionado ha sido revisado y se han añadido la determinación de LDH junto con la presencia o ausencia de alteraciones citogenéticas para identificar de nuevo a tres grupos de pacientes con diferente valor pronóstico: estadio R-ISS I, cuando los pacientes son ISS 1 con citogenética y LDH normales o estadio R-ISS III, cuando el ISS es 3 junto con, citogenética de alto riesgo y/o LDH elevadas y estadio R-ISS II cuando no cumple los criterios de I ni III.
¿Cuál es el pronóstico de la enfermedad?
El Mieloma Múltiple es una enfermedad compleja y es dificil generalizar. Desde el punto de vista general, el Mieloma es una enfermedad incurable. Sin embargo, el pronóstico de la enfermedad no es homogéneo y, como se ha mencionado anteriormente, existen diferentes sistemas de estadiaje de la enfermedad que permiten definir el estadio de la enfermedad en cada paciente y que asocia un pronóstico determinado. Tanto el sistema ISS como el nuevo R-ISS son válidos para establecer el pronóstico de la enfermedad.
Las alteraciones cromosómicas son importantes factores pronósticos y, de hecho, las opciones de tratamiento podrían variar en función de su presencia o no. Todos estos factores pronósticos se evalúan en el momento del diagnóstico de la enfermedad. Sin embargo, durante la evolución, una vez se inicie el tratamiento, la respuesta al mismo es uno de los fatores pronósticos más importantes y se sabe que la mejor calidad de la respuesta se asocia, en general, con una supervivencia más prolongada. En este sentido, sus médicos le pueden pedir la realización de un nuevo aspirado de médula ósea durante el tratamiento, cuando el componente monoclonal haya desaparecido en el suero y que se conoce como respuesta completa.
El objetivo será confirmar que las células plasmáticas del mieloma han desaparecido de la médula pero además, se le podrá evaluar la enfermedad mínima residual que consiste en detectar una célula plasmática del Mieloma que permanezca entre al menos 100.000 o incluso 1.000.000 de células normales. Cuando su enfermedad mínima residual sea negativa, la posibilidad de que la enfermedad aparezca de nuevo disminuye de una manera muy significativa adicional para intentar erradicarla.
¿Cómo se vive con un Mieloma Múltiple?
Cuando un paciente es diagnosticado de Mieloma Múltiple, el primer aspecto importante y a veces desalentador es entender una enfermedad desconocida y que puede parecer complicada. Este debe ser el primer paso: conocer la enfermedad y entenderla. En segundo lugar, saber cómo se diagnostica, las pruebas necesarias e incluso cuál es el estadio de la enfermedad. El paciente tiene que conocer las opciones de tratamiento inicial en el momento del diagnóstico y como se va a evalúar la eficacia del tratamiento.
Por último tiene que saber que existe tratamiento de soporte para aliviar la sintomatología física así como la carga emocional que supone el diagnóstico de la enfermedad. Estas medidas son tan importantes como el tratamiento de primera línea y el paciente con Mieloma puede contar con medidas de apoyo como son: 1) la actividad física moderada y adaptada a la enfermedad ósea que presenten, 2) hacer una dieta sana evitando la toma de suplementos de hierbas o vitaminas sin consultarlo con su médico, 3) la salud mental requiriendo apoyo psicológico si cree que pueda presentar algún síntoma de depresión, 4) tener un sueño regular ya que es importante para el sistema inmunitario, 5) ajustes en la vida para intentar minimizar el estrés en la familia, trabajo y entorno social. El paciente también tiene que preguntar a su médico cuestiones importantes y relevantes acerca de como proceder con su vida cotidiana, sus actividades diarias, su trabajo, los efectos secundarios que presentará, e incluso que sucederá si el tratamiento inicial no funciona, las opciones siguientes de tratamiento e incluso sobre la evolución de la enfermedad a largo plazo.
Una vez el paciente reciba toda esta información, la tiene que asimilar y “digerir” para intentar mantener la vida diaria lo más normal posible lo cual será más sencillo si la enfermedad responde bien al tratamiento.
Además, los pacientes tienen que saber que existen asociaciones de pacientes, con cáncer en general y Mieloma en particular, que pueden servirle de ayuda y soporte tanto al paciente como a su familia en las fases iniciales con la posibilidad de convertirse en un miembro activo de las mismas contribuyendo a ayudar en un futuro a otras personas con Mieloma.
Leucemia Linfocítica Crónica
¿Qué es la Leucemia Linfocítica Crónica?
La leucemia linfocítica crónica (que también se conoce como LLC por sus siglas) es un tipo de cáncer en el que la médula ósea produce demasiados linfocitos B (un tipo de glóbulo blanco) y puede afectar la producción de otras células de la sangre como los glóbulos rojos, glóbulos blancos y plaquetas. Hay varios tipos de linfocitos, cuya proliferación incontrolada puede dar lugar a distintos tipos de leucemia. Sin embargo, el tipo de leucemia más frecuente con diferencia es la que afecta a los linfocitos B, y es a la que nos referiremos a lo largo de esta guía (simplemente como LLC).
La LLC es un tipo de enfermedad maligna de la sangre y del lugar donde ésta se fabrica, la médula ósea (tuétano) y que tiende a producir adenopatías (inflamación de los ganglios linfáticos). La LLC es el tipo más común de leucemia en adultos, se suele presentar durante o después de la edad madura y la frecuencia va aumentando con la edad. La mayoría de los enfermos se diagnostican con más de 70 años, aunque puede ocurrir a edades más jóvenes.


Se llama leucemia linfocítica crónica para diferenciarla de la leucemia linfoblástica aguda:

Normalmente, el cuerpo produce células madre sanguíneas (células inmaduras) que se convierten, con el tiempo, en células sanguíneas maduras. Una célula madre sanguínea se puede convertir en una célula madre mieloide (que dará lugar, entre otros, a glóbulos rojos, plaquetas y algunos tipos de leucocitos) o una célula madre linfoide (que dará lugar a los linfocitos B y T).
En el caso de la LLC hay demasiadas células madre sanguíneas que se convierten en linfocitos B anormales, que no combaten muy bien las infecciones. Además, a medida que aumenta el número de linfocitos en la sangre y la médula ósea, hay menos lugar para los glóbulos blancos, los glóbulos rojos y plaquetas. Esto puede producir infecciones, anemia y sangrados.
¿Qué causa la Leucemia Linfocítica Crónica?
No se sabe cuál es la causa que produce la leucemia linfocítica crónica. A diferencia de otros tipos de leucemia, no se ha comprobado que la exposición a radiaciones o a otros tóxicos aumenten el riesgo de sufrir la enfermedad.
A pesar que está pueda tener algunas alteraciones en los genes de las células tumorales, eso no quiere decir que la enfermedad se transmita de padres a hijos. Esas alteraciones genéticas o moleculares se adquieren en algún momento a lo largo de la vida y pueden asociarse con el desarrollo de la enfermedad. Se pueden encontrar o no al diagnóstico y durante los años de evolución de la enfermedad pueden ir apareciendo nuevas alteraciones, con frecuencia favorecidas por alguno de los tratamientos recibidos.
Por otra parte, en algunas familias hay mayor predisposición a lo habitual de sufrir leucemia linfocítica crónica. Aún en esas raras situaciones, no se trata de una herencia definida como en las verdaderas enfermedades hereditarias, simplemente se trata de una mayor tendencia a desarrollarla en comparación con lo esperable.
Síntomas de la Leucemia Linfocítica Crónica
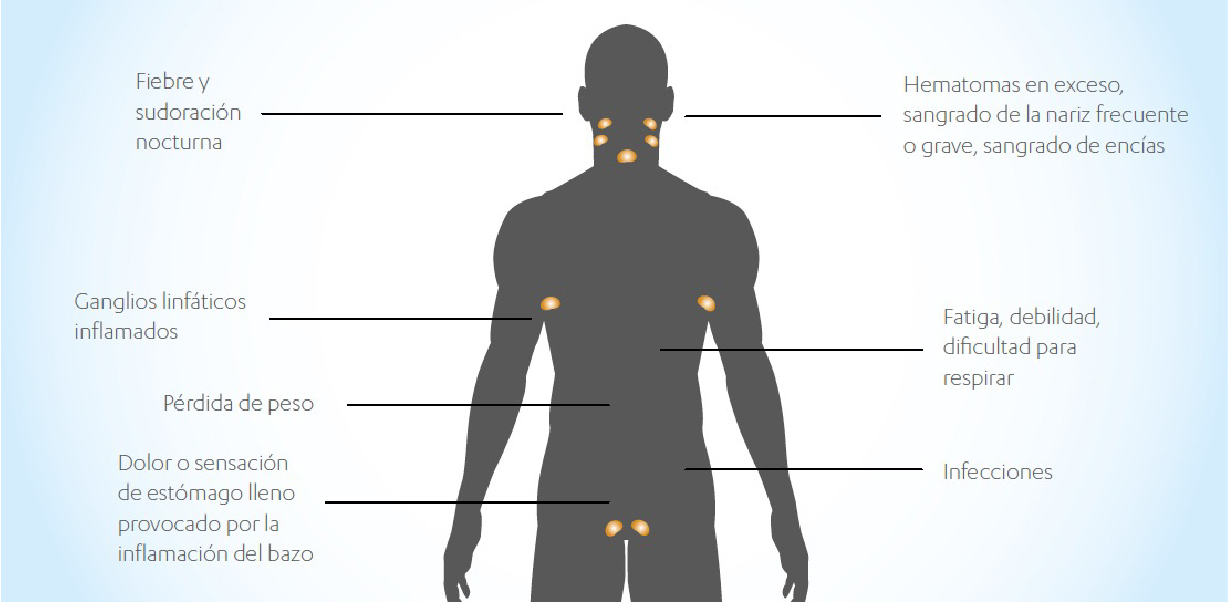
En la mayoría de casos, la LLC no presenta signos ni síntomas. Usualmente se detecta en un análisis de sangre de rutina. Algunas veces se presentan signos o síntomas que pueden ser producidos por la LLC, pero ninguno de ellos son específicos, pudiendo ser causados por otros problemas médicos. Consulte a su médico si presenta, de forma continua en el tiempo, cualquiera de los siguientes problemas:
- Hinchazón sin dolor de los ganglios linfáticos en cuello, axilas o ingles.
- Mucho cansancio.
- Dolor o sensación de ocupación debajo de las costillas.
- Fiebre de forma continuada, sobre todo por las tardes.
- Sudoración marcada sobre todo por las noches.
- Pérdida de peso llamativa sin razón aparente.
Diagnóstico de la Leucemia Linfocítica Crónica
Entre las pruebas y los procedimientos que se utilizan para diagnosticar la leucemia linfocítica crónica se encuentran los siguientes:
- Examen físico : se valora la presencia de ganglios linfáticos aumentados de tamaño (adenomegalias o adenopatías), aumento de tamaño del hígado o del bazo, masas o cualquier otra cosa que pueda ser anormal. La exploración no sirve para el diagnóstico pero sí para valorar el estadio de la enfermedad (ver más adelante).
- Recuento sanguíneo completo: en una muestra de sangre un médico al microscopio confirma un exceso de linfocitos que son de tamaño pequeño, aspecto maduro y que se rompen con facilidad. Una LLC deberá tener más de 5000 linfocitos por mm3, que tengan determinadas características (inmunofenotipo, ver a continuación).
- Inmunofenotipo por Citometría de flujo: prueba fundamental, es realmente la que confirma el diagnóstico. Se examinan los marcadores o antígenos en la superficie de los linfocitos de la sangre. Si estas células son linfocitos malignos (cancerosos) se suelen poder diferenciar bien con esta técnica de otras enfermedades linfoproliferativas. No siempre es sencillo diferenciar casos no típicos de LLC de otras enfermedades malignas de linfocitos B, como linfomas de células del manto.
- FISH (por las siglas en inglés de hibridación fluorescente in situ): técnica de laboratorio que se usa para encontrar, por medio de un microscopio de luz fluorescente, determinadas alteraciones cromosomicas. Esta técnica es importante para definir el tipo de tratamiento cuando éste sea necesario, no se requiere para el diagnóstico.
- Aspiración y biopsia de la médula ósea: extracción de médula ósea, sangre y un trozo pequeño de hueso mediante la inserción de una aguja hueca en el hueso de la cadera. No es necesaria para el diagnóstico, ya que con la sangre se pueden hacer las pruebas mencionadas. Sí que puede ser útil para estudiar casos de LLC con anemia o plaquetas bajas, para diferenciar si esas situaciones se deben a la ocupación de la médula por la LLC o bien por un mecanismo autoinmune (mala regulación de las defensas propias que destruyen de forma acelerada glóbulos rojos o plaquetas). Otra utilidad de la biopsia es confirmar una respuesta aparentemente completa tras haber recibido tratamiento.
- Estadios: Desde hace décadas, se utilizan sistemas para establecer el pronóstico (predecir la evolución mejor o peor de la enfermedad) basados en los recuentos sanguíneos y en la exploración física. De forma abreviada, un estadio 0 o A supone tener linfocitosis, sin adenopatías o con pocas adenopatías significativas, estadio I-II o B tienen más adenopatías o crecimiento de hígado/bazo y los estadios III-IV o C serían aquellos con anemia o plaquetas bajas por infiltración de la enfermedad.
Pronóstico de la Leucemia Linfocítica Crónica
Se entiende por pronóstico la predicción respecto al futuro desarrollo de la salud del paciente, cómo se espera que vayan las cosas en el tiempo que se tiene por delante, qué probabilidades existen de estar vivo a largo plazo o de estar sin progresión de la enfermedad, que posibilidades se esperan de que determinado tratamiento sea beneficioso, etc. Hay múltiples factores que tienen importancia, como los siguientes:
- Estadio clínico de la enfermedad: como vimos anteriormente, estadios avanzados implican anemia o plaquetas significativamente bajas debido a la LLC.
- Si se encuentran o no determinadas alteraciones genéticas o moleculares, como hemos visto previamente. Especialmente la pérdida o mutación del gen p53, que se encuentra en el cromosoma 17, puede hacer cambiar la decisión de tratamiento de su médico, porque en esos casos no es esperable benefício con algunas opciones.
- Si la LLC mejora o no con el tratamiento recibido (se dice que se alcanza respuesta, que puede ser completa o parcial).
- Si la LLC, en casos de recaída, tuvo un largo período de duración de la respuesta previa o no.
- Si la LLC evoluciona a una enfermedad maligna linfoide más agresiva, como linfomas (síndrome de Richter) o leucemia prolinfocítica.
- La salud general del paciente. Un mejor estado general y una menor cantidad de otros problemas médicos asociados, siempre van a suponer mejor pronóstico.
La LLC es una enfermedad que puede tener una evolución muy variable entre unos pacientes y otros, incluso entre los que compartan los mismos factores pronósticos. Es conveniente tener esto en cuenta, porque la evolución a largo plazo puede no ser fácil de predecir.
¿Cómo se vive con Leucemia Linfocítica Crónica?
Muchos de los pacientes recién diagnosticados no necesitan recibir ningún tratamiento al ser diagnosticados, su médico mantendrá vigilancia de la enfermedad con revisiones periódicas, esperando a que existan datos que supongan una progresión significativa de la enfermedad. Fuera de estudios de investigación, no se recomienda iniciar tratamiento hasta que esa clara progresión ocurra. Su médico al plantearle tratamiento se basará en una de las siguientes situaciones:
- Inflamación muy voluminosa o progresiva de los ganglios linfáticos (adenopatías) o crecimiento del hígado o del bazo.
- Anemia o plaquetas bajas, de forma significativa (en general con menos de 10-11 g/dl de hemoglobina, y menos de 100.000 plaquetas que van disminuyendo progresivamente, que sean atribuibles a la ocupación de la médula. Si fuera por autoinmunidad, se usarían tratamientos inmunosupresores y sólo sería planteable pensar en tratar la LLC si fallaran esos inmunosupresores.
- Síntomas generales atribuibles a la LLC: lo mencionado antes, destacando que siempre se descartarán otras posibles causas (infecciones, problemas inflamatorios, otros tumores, etc.):
- Mucho cansancio.
- Fiebre (o “décimas”) de forma continuada, sobre todo por las tardes.
- Sudoración marcada sobre todo por las noches.
- Pérdida de peso llamativa sin razón aparente.
Es importante saber que las mismas razones se aplican para iniciar un primer tratamiento que para las posibles siguientes terapias, tras eventuales reapariciones de la enfermedad. En los períodos de vigilancia sin tratamiento es frecuente que se pueda sentir ansiedad o preocupación ante lo incierto del futuro, pero su médico le podrá explicar cómo debe tratar de mantener una mejor calidad de vida posible, sin pensar en la enfermedad ni limitar innecesariamente sus actividades habituales.
Referencia bibliográfica
- Loscertales Pueyo, Javier; Font López, Patricia; Jiménez Barral, Elena; Acuña Cruz, Evelyn. Tratamiento de la leucemia linfocítica crónica y otros síndromes linfoproliferativos crónicos. Capítulo 11 del Libro “Terapia en Oncohematología”, 4ª edición. Aymon solutions, 2018.
- García-Marco et al. Actualización de las guías nacionales de consenso del Grupo Español de LLC para el tratamiento de la leucemia linfocítica crónica. En prensa, 2018.
Linfoma de Células del Manto
¿Qué es el linfoma de células del manto?
El linfoma de células del manto es un tumor del sistema linfático y se debe a la acumulación y proliferación descontrolada de un tipo de glóbulos blancos o leucocitos llamados “linfocitos B”, que se transforman en células malignas o cancerígenas. Existen unos linfocitos B que viven en una zona del ganglio linfático llamada “zona del manto folicular”. El carácter de malignidad de este cáncer se reconoce porque los linfocitos B tumorales se multiplican, extendiéndose y rebasando en el ganglio linfático la zona en la que las células normales del manto folicular residen en condiciones normales. Los linfocitos B del linfoma del manto se dividen descontroladamente, produciendo un aumento de tamaño de los ganglios linfáticos. Si se trata de ganglios superficiales bajo la piel, es fácil que se detecten como tumoraciones o “bultos” a los lados de cuello, axilas o ingles. Si los que crecen son ganglios internos, pueden tardar en detectarse, hasta que produzcan síntomas. Las células tumorales pueden salir de los ganglios linfáticos, y detectarse por lo tanto, en un análisis de sangre, y pueden localizarse y proliferar a distancia en otros ganglios linfáticos o tejidos como la médula ósea, el hígado, el bazo y otros sistemas, como el sistema digestivo, respiratorio o nervioso central.

El linfoma de células del manto es un tipo de linfoma poco frecuente, ya que representa cerca del 5% de todos los linfomas. La edad de presentación es en general por encima de los 60 años, pero puede aparecer desde los 35 años en adelante. es dos veces más frecuente en hombres que en mujeres y cuando se diagnostica suele ser en estadios avanzados (›90%) 1. A la presentación del linfoma, más del 80% de los pacientes tienen síntomas de enfermedad, y en un 40% de casos los síntomas incluyen fiebre (›38º), sudores nocturnos y pérdida de peso. A estos tres síntomas es lo que se conocen como “síntomas B”. En menos del 10% de los pacientes se presentan astenia (cansancio), anorexia (falta de apetito), malestar general o prurito. Otro tipo de síntomas locales, como cefalea, tos, dificultad para respirar, dolor óseo o abdominal pueden indicar afectación a esos niveles de la enfermedad 2. Lo habitual es encontrar adenopatías en diversos territorios ganglionares, aumento del tamaño del hígado y bazo. Es frecuente la anemia y la afectación de la médula ósea por el linfoma. Menos frecuente, pero se ha de buscar porque puede pasar desapercibido, es la afectación extraganglionar, es decir, afectación de otros órganos que no sean los ganglios linfáticos, como puede ser el tracto digestivo o respiratorio, y más raramente el sistema nervioso, mama o glándulas salivares
¿Qué causa el Linfoma de Células del Manto?
La causa por la que se produce este tipo de linfoma es desconocida. Todos los linfomas se originan de células de nuestro sistema inmunológico llamados “linfocitos”.
A modo de resumen, lo que suele ser característico en el linfoma de células de manto es la traslocación t(11;14), es decir, entre el cromosoma 11 y el cromosoma 14.
Síntomas del Linfoma de Células del Manto
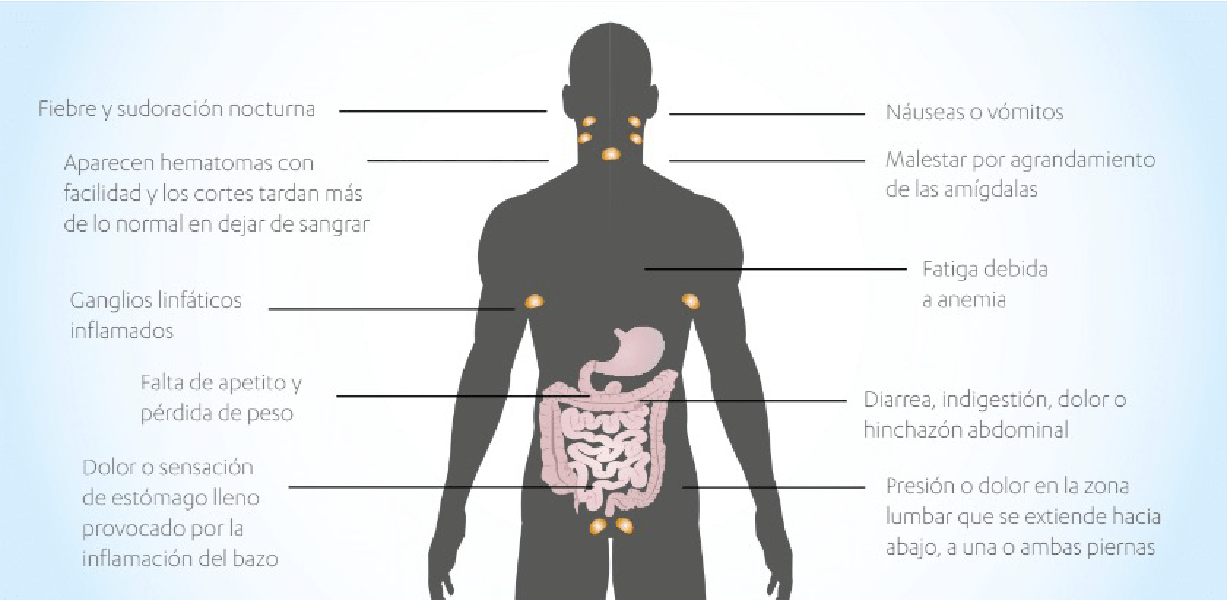
La mayoría de los pacientes con linfoma de células del manto tienen una enfermedad que afecta a múltiples lugares del cuerpo, principalmente a los ganglios linfáticos.
Otras localizaciones de este linfoma pueden incluir el bazo, la médula ósea y la sangre, el tejido linfático de la garganta (amígdalas y adenoides), el hígado, el sistema gastrointestinal o el aparato respiratorio. Las células del linfoma del manto también pueden entrar en el cerebro y la médula espinal, lo que es relativamente raro pero potencialmente peligroso porque requiere entonces un tratamiento diferente, que pueda atravesar la barrera que protege al sistema nervioso central. Un pequeño porcentaje, menor al 5%, de los pacientes con linfoma de células del manto pueden encontrarse libres de síntomas al diagnóstico, si éste se hace por un análisis de sangre o al estudiar un pequeño bulto como un aumento del tamaño del bazo, y en estos casos su comportamiento puede ser más benigno y no requerir tratamiento. Pero la mayoría de los pacientes suelen encontrarse enfermos por esta causa. Los síntomas que pueden deberse al linfoma del manto suelen ser la falta de apetito y pérdida de peso, fiebre, sudoración nocturna, náuseas o vómitos, malas digestiones, dolor abdominal o hinchazón, sensación de “saciedad” o molestias de garganta por el agrandamiento de las amígdalas. Los ganglios linfáticos aumentados de tamaño pueden ser molestos, pero en general no son dolorosos. El hígado o el bazo aumentados de tamaño pueden producir los síntomas abdominales anteriores, pero también, y al igual que los ganglios linfáticos abdominales si son muy grandes, pueden provocar presión o dolor en la zona lumbar, que puede extenderse a una o ambas piernas.
Si existe anemia, puede manifestarse como fatiga y palidez o debilidad ante actividades que suponen pequeños esfuerzos. Si bajan las plaquetas, también llamado trombocitopenia, puede manifestarse como hemorragias fáciles en forma de hematomas, pequeños puntos rojos superficiales en la piel llamados petequias, o hemorragias digestivas. La anemia y trombocitopenia son debidas a que las células del linfoma ocupan la médula ósea e impiden la producción de células sanguíneas. Si es así, además se puede detectar la presencia en la sangre de las células del linfoma, con un aumento de los recuentos de linfocitos, lo que se denomina la fase leucémica de la enfermedad. Según avanza la progresión del linfoma, se multiplican las manifestaciones de la enfermedad. Debido a que el linfoma de células del manto produce con mucha frecuencia afectación extraganglionar, es común que se presenten síntomas fuera de los ganglios y otros órganos linfáticos. En el tubo gastrointestinal, pueden desarrollarse múltiples “pólipos” en el intestino delgado y grueso, por la extensión y proliferación de las células del linfoma por debajo de la mucosa intestinal, que pueden asociarse a grandes masas de linfoma en territorios adyacentes. También el linfoma del manto se puede extender a la mucosa gastrointestinal formando úlceras, que en ocasiones producen hemorragias digestivas.
RECONOCER UNA INFECCIÓN
Aprender a identificar los síntomas de una infección es muy importante para su autocuidado. Estos son algunos síntomas que podrían indicar que está desarrollando una infección:
- Sensación de calor/temperatura corporal mayor o igual a 38°C
- Escalofríos/sudoración
- Tos, dolor de garganta o sensación de que le falta el aire
- Dolor de oídos
- Dolor de cabeza
- Rigidez o dolor en el cuello
- Llagas o puntos blancos en la boca o la lengua
- Enrojecimiento o inflamación
- Orina turbia o con sangre, molestias al orinar
- Secreción o irritación vaginal
Diagnóstico del Linfoma de Células del Manto
Cuando un paciente se enfrenta al posible diagnóstico de linfoma debe tener presente que el médico especialista, habitualmente un hematólogo-oncólogo, y el equipo multidisciplinario que le apoya realizará las pruebas necesarias para asegurarse de que el diagnóstico es de linfoma, descartando otras reacciones del sistema linfático e inmunitario frente a otros procesos de carácter infeccioso, inflamatorio, degenerativo o de otra naturaleza tumoral.
Al tiempo, una vez realizado el diagnóstico de linfoma, es necesario identificar correctamente la clase de linfoma de que se trata y el subtipo reconocido. Estos pasos son necesarios para establecer las pruebas que hay que realizar en cada paciente, determinar el pronóstico y seleccionar el tratamiento más adecuado, puesto que cada subtipo de linfoma tiene unas opciones de tratamiento específicas.Para realizar el diagnóstico de un linfoma es necesario siempre el análisis del tejido afectado por el tumor, por lo que es obligatoria la realización de una biopsia. Habitualmente, el diagnóstico se efectúa con el análisis microscópico y la ayuda de técnicas moleculares del material obtenido por una biopsia quirúrgica. La mayoría de los linfomas se diagnostican por la biopsia de un ganglio linfático, pero también puede ser la biopsia de cualquier otro órgano afectado por el linfoma.
El diagnóstico de los linfomas requiere una biopsia del ganglio o del órgano afectado y un examen histopatológico. La biopsia ganglionar se trata de una intervención quirúrgica, por lo que el paciente debe someterse a una valoración de los riesgos de la cirugía o de la anestesia.
El patólogo, el médico especialista que realiza el análisis microscópico, tiene que procesar la biopsia, para posteriormente poder trabajar con ella y realizar tinciones que permitan establecer el diagnóstico. Todo este proceso puede tener una duración variable, habitualmente entre 7 y 14 días, aunque en algunos casos complicados el proceso diagnóstico puede prolongarse.
Además de la biopsia, se realizarán pruebas de imagen para averiguar cuántos órganos se encuentran afectos por el linfoma. Para ello se realizan técnicas de Tomografía Axial Computarizada (TAC) o también llamado “escaner” o bien técnicas de Tomografía de Emisión de Positrones (PET).
Como puede estar afectada la médula ósea, es necesario siempre realizar una biopsia de la médula ósea que puede hacerse en la cresta iliaca posterior o en el esternón. Otras pruebas adicionales pueden incluir, punciones lumbares para el análisis del líquido cefalorraquídeo, exploraciones por el otorrinolaringólogo y endoscopias digestivas (gastroscopia y colonoscopia), para comprobar si existe afectación por linfoma de estas áreas.
¿QUÉ ES LA ESTADIFICACIÓN DEL LINFOMA?
La estadificación es el conjunto de procedimientos que permite determinar con precisión la extensión de la enfermedad, es decir, qué órganos y sistemas del organismo están afectados por el linfoma. La clasificación de los linfomas por estadios de extensión permite a los médicos establecer el pronóstico (predicción del curso futuro de la enfermedad y las posibilidades de necesidad y respuesta al tratamiento y la supervivencia). Según los estadios se pueden adaptar los tratamientos para cada paciente en particular, lo que junto a otros factores dependientes del paciente como son la edad, la presencia de otras enfermedades o antecedentes médicos y la fragilidad estimada, le orientan al médico a seleccionar el tratamiento más conveniente para conseguir resultados positivos y reducir los posibles efectos tóxicos o negativos de la terapia. También hay un índice pronóstico específico del linfoma de células del manto que es el MIPI (del inglés, Índice Pronóstico Internacional del Linfoma de células del manto), que es predictor únicamente de la supervivencia. El MIPI tiene en cuenta cuatro factores en el momento del diagnóstico que son independientes entre sí: edad; estado general (capacidad de realizar actividades de la vida diaria); nivel de deshidrogenasa láctica (LDH) en la sangre; y el recuento de leucocitos (glóbulos blancos) en la sangre.
Pronóstico del Linfoma de Células del Manto
Se han logrado avances extraordinarios en el tratamiento del linfoma de células del manto durante las últimas décadas, aunque las recaídas siguen siendo un problema frecuente, que suceden en la inmensa mayoría de los pacientes.
El pronóstico para la variante blástica del linfoma de células del manto sigue siendo el menos favorable. Los investigadores continúan buscando terapias que puedan prolongar las remisiones y que extiendan el beneficio en los pacientes. Estas terapias se analizan dentro de “ensayos clínicos”, que es la forma que se tiene en medicina de demostrar que una nueva terapia es eficaz y segura para el paciente.
¿Cómo se vive con Linfoma de Células del Manto?
Los pacientes jóvenes van a tener que acudir muy frecuentemente al hospital y van a precisar ingresos hospitalarios prolongados, lo cual va a interferir en su calidad de vida y en su actividad laboral y profesional, durante al menos 6 meses.
Los pacientes jóvenes van a tener que acudir muy frecuentemente al hospital y van a precisar ingresos hospitalarios, lo cual va a interferir en su calidad de vida y en su actividad laboral y profesional, durante al menos 6 meses.
A los pacientes mayores no se les puede ofrecer tratamientos tan intensivos como a los de menor edad. En función de su estado general, calidad de vida, la presencia de otras enfermedades o la detección de fragilidad o vulnerabilidad, hace que tengamos que adaptar la intensidad de los tratamientos de forma individualizada en cada paciente.
En la actualidad se están produciendo grandes avances en el conocimiento de esta patología, lo que ha conseguido mejorar la calidad de vida de estos pacientes.
Referencia bibliográfica
- McKay, P., Leach, M., et al. (2012). Br J Haematol. 159(4): 405-26.
- Williams, M.E., Bernstein, S.H., et al. (2013). Leuk Lymphoma.
- Cortelazzo et al. 2012. Crit Rev Oncol Hematol. 82(1): 78-101.
- *El CD20 es un antígeno del sistema inmunitario que se expresa en las células B y puede servir como marcador en algunos cánceres, como por ejemplo, los linfomas de células B y las leucemias.
Leucemia Mieloide Aguda
¿Qué es la Leucemia Mieloide Aguda?
El término leucemia abarca una serie de enfermedades que afectan al sistema hematopoyético, responsable de la fabricación de los elementos formes (células) normales que circulan en la sangre.
La leucemia mieloide aguda es un tipo de cáncer por el que la médula ósea produce, de manera anormal, mieloblastos (un tipo de glóbulo blanco muy inmaduro).
La leucemia puede afectar los glóbulos rojos, los glóbulos blancos y las plaquetas. Cuando la hematopoyesis (proceso de formación, desarrollo y maduración de las células de la sangre) es normal, las células precursoras maduran y se transforman en células sanguíneas; en cambio, cuando existe leucemia mieloide, hay un paro madurativo, se acumulan precursores sanguíneos de la estirpe mieloide en la medula ósea (llamados blastos o mieloblastos), lo cual imposibilita que se formen células sanguíneas funcionales (glóbulos rojos, glóbulos blancos y plaquetas).
Las células anormales y disfuncionales reemplazan a las maduras y funcionales, lo que acarrea, entre otros problemas, una disminución de las defensas frente a infecciones. Estas alteraciones en las series sanguíneas pueden verse en un análisis de sangre (hemograma).
La leucemia mieloide aguda (LMA) se caracteriza por una transformación muy rápida y repentina que se manifiesta en una producción veloz de numerosos glóbulos blancos, en su mayoría inmaduros. Esta situación supone una amenaza para la vida y debe tratarse de forma inmediata.
La LMA es el resultado de cambios (mutaciones) adquiridos en el ADN (material genético) de una célula de la médula ósea en desarrollo. Una vez que la célula de la médula ósea se transforma en una célula leucémica, se multiplica en gran proporción. Estas células, llamadas “blastos leucémicos”, no funcionan normalmente. Sin embargo, crecen y sobreviven mejor que las células normales.
La presencia de blastos leucémicos impide la producción de células normales. Como resultado, cuando se diagnostica un caso de LMA, la cantidad de células sanguíneas sanas (glóbulos rojos, glóbulos blancos y plaquetas) suele ser menor de la normal.
¿Qué causa la Leucemia Mieloide Aguda?
Ciertos cambios en el ADN pueden causar que las células normales de la médula ósea se conviertan en células de leucemia. Las células humanas normales crecen y funcionan basándose en la información contenida en los cromosomas de cada célula.
Hay ciertos genes que ayudan a las células a crecer, dividirse y a vivir más tiempo, se les denomina oncogenes. Otros, enlentecen la división celular o causan que las células mueran en el momento oportuno, se les llama genes supresores de tumores.
Las mutaciones en genes específicos que controlan la división celular se encuentran en muchos casos de LMA, aunque también son comunes cambios mayores en uno o más cromosomas.
En la mayoría de los casos no es posible encontrar una causa clara para la aparición de la LMA, podrían ser causa de LMA las siguientes:
- Enfermedades sanguíneas en el pasado (p. ej. síndrome mielodisplásico – SMD)
- Tratamiento con citostáticos (medicamentos que se administran en el marco de una quimioterapia y que influyen en la división celular)
- Radioterapia
- Contacto con tóxicos como el benceno durante varios años (presente, por ejemplo, en algunos pesticidas)
- Tabaco
- Trastornos genéticos, tales como la anemia de Fanconi, el síndrome de Shwachman, el síndrome de Diamond-Blackfan y el síndrome de Down, se asocian con un aumento del riesgo de presentar LMA.
Síntomas de la Leucemia Mieloide Aguda
Los signos y síntomas de la LMA son muy inespecíficos y eso puede suponer retrasos en el diagnóstico.
Es común que las personas con LMA sientan una pérdida de bienestar, debido a la producción insuficiente de células normales de médula ósea. Es posible que la persona se canse con más frecuencia y que le falte el aliento durante las actividades físicas normales debido a una baja cifra de glóbulos rojos.
A continuación se presentan los síntomas típicos de la LMA:
- Mayor propensión a las infecciones (resfriados, gripe, etc.)
- Hemorragias
- Cansancio, agotamiento
- Fiebre
- Sudores nocturnos
- Pérdida de peso
- Dolor de huesos o articulaciones
- Inflamación de los ganglios linfáticos
- Tos irritativa
- Puede que las personas con LMA tengan además:
- Palidez a causa de la anemia
- Signos de sangrado causado por un recuento muy bajo de plaquetas, como cardenales o hematomas sin motivo aparente o debidos a una lesión menor
- Aparición en la piel de puntos rojos, del tamaño de una cabeza de alfiler, llamados “petequias”
- Sangrado prolongado por cortaduras leves
- Encías inflamadas
- Infecciones menores frecuentes, tales como llagas perianales (en el área alrededor del ano)
- Pérdida de apetito y pérdida de peso
- Aumento del tamaño del bazo
- Aumento del tamaño del hígado
- Sarcoma mieloide
Diagnóstico de la Leucemia Mieloide Aguda
Es importante obtener un diagnóstico preciso del tipo de leucemia que se padece. El diagnóstico exacto le ayuda al médico a:
- Estimar cómo progresará la enfermedad.
- Determinar el tratamiento adecuado.
Algunas de las pruebas que se describen a continuación pueden repetirse durante y después del tratamiento para medir los efectos del mismo.
Pruebas de sangre y médula ósea. Las pruebas de sangre y médula ósea se usan para diagnosticar la LMA y el subtipo de la LMA. Un cambio en la cantidad y la apariencia de las células sanguíneas ayuda a hacer el diagnóstico. Las células leucémicas en el microscopio se parecen a los glóbulos blancos inmaduros normales. Sin embargo, su desarrollo está incompleto.
Muestras de sangre y médula ósea. Para hacer las pruebas, se suelen tomar muestras de sangre de una vena del brazo del paciente. Las muestras de células de la médula ósea se obtienen mediante una aspiración y una biopsia de médula ósea.
Pruebas de sangre que se usan según qué tenga el paciente:
Recuentos de glóbulos rojos y plaquetas menores de lo habitual: CBC: los recuentos de células sanguíneas se determinan mediante una prueba de sangre llamada “hemograma completo” (CBC, por sus siglas en inglés).
Exceso de glóbulos blancos inmaduros e insuficientes glóbulos blancos maduros: Frotis de sangre periférica. Consiste en un examen visual de las células sanguíneas coloreadas o teñidas que se realiza con un microscopio.
Confirmación del diagnóstico. Además de observar la cantidad y la apariencia de las células de las muestras de sangre, su médico pedirá que le realicen otras pruebas para:
- Confirmar el diagnóstico
- Identificar el subtipo de la LMA
- Desarrollar un plan de tratamiento
Su médico habitual le derivará a un hematólogo para confirmar el diagnóstico. Un hematólogo es un especialista encargado del diagnóstico, tratamiento y seguimiento de las enfermedades relacionadas con las sangre y los órganos que la producen. El diagnóstico de LMA se confirma identificando células blásticas leucémicas en muestras de médula ósea.
Los blastos suelen constituir en condiciones normales entre el 1 y el 5% de las células de médula ósea. Por lo general se necesita tener al menos un 20% de blastos para hacer un diagnóstico de LMA. Existen algunos tipos de LMA que se pueden diagnosticar si los blastos tienen un cambio cromosómico que ocurre en un tipo específico de LMA, aun cuando el porcentaje de blastos sea menor de 20%.
Actividad química específica en las células blásticas.
Marcadores característicos (antígenos) sobre la superficie de las células blásticas, tales como CD13 o CD33
Citometría de flujo, prueba que permite hacer el inmuno-fenotipado (estudio de los tipos de marcadores (antígenos) en la superficie de la célula).
Otras pruebas:
El estudio citogenético a través del cariotipo, de la FISH (hibridación in situ fluorescente, por sus siglas en inglés) y de la biología molecular (técnicas de PCR), se usa para identificar ciertos cambios estructurales o funcionales en los cromosomas, e incluso concretamente en algunos genes.
LEYENDA
inv= inversión¥. LA= leucemia aguda. LMA= leucemia mieloide aguda. NOS= not otherwise specified (no especificado de otro modo). p= brazo corto del cromosoma. q= brazo largo del cromosoma. t= traslocación+.
¥Inversión: Una inversión implica que un segmento del cromosoma cambia de lugar dentro del mismo cromosoma.
+traslocación: Una translocación es el desplazamiento de un segmento de un cromosoma a otro cromosoma, como por ejemplo que parte del cromosoma 8 se localice en el 21, se indica como t(8;21)
Pronóstico de la Leucemia Mieloide Aguda
El pronóstico de los pacientes diagnosticados con leucemia mieloide aguda varía sustancialmente en función de la edad del paciente y del subtipo de la LMA. La edad avanzada, las LMA relacionadas con tratamientos previos, o secundarias a una mielodisplasia o síndrome mieloproliferativo, el grado de leucocitosis inicial (aumento de leucocitos circulantes en la sangre), la presencia de determinadas anomalías genéticas/moleculares, así como la lentitud en la obtención de la respuesta al tratamiento entre otros, constituyen parámetros de pronóstico desfavorable.
Así, los pacientes jóvenes con leucemias de riesgo estándar que reciben un trasplante de médula alogénico familiar o de donante no emparentado en primera remisión completa tienen una probabilidad de curación de hasta el 65%, mientras que un paciente de edad avanzada, con una leucemia post-mielodisplásica o secundaria que no alcance la remisión completa con la quimioterapia de inducción, tienes menos opciones de tratamiento curativo.
¿Cómo se vive con Leucemia Mieloide Aguda?
El tratamiento de la leucemia mieloide aguda varía dependiendo de muchos factores. Uno de los principales es la edad biológica, es decir, la edad que corresponde al estado físico y a la salud del paciente.
Múltiples facetas de la vida diaria como la alimentación, nivel de actividad física y estado mental pueden afectar al estado de salud por lo que es muy importante mantener buenos hábitos en dichos ámbitos.
Nutrición
La nutrición es uno de los factores que deben cuidar las personas que están recibiendo un tratamiento contra la leucemia mieloide aguda. La dieta debe ser equilibrada, el hecho de incluir en ella alimentos indicados por los profesionales, durante el desarrollo del tratamiento y cuando éste finaliza, puede suponer una mejora muy notable haciendo que el paciente se sienta con más energía.
Los efectos secundarios generados durante el tratamiento (vómitos, náuseas, cambios en el sentido del gusto…) pueden provocar pérdida de apetito y esto conlleva una pérdida de peso involuntaria. Para intentar sobrellevar estos síntomas se aconseja comer porciones pequeñas cada dos o tres horas, estos problemas suelen remitir con el paso del tiempo.
Posteriormente, una vez remitidos los síntomas iniciales, aproximadamente un 60% de las personas diagnosticadas de cáncer aumentan de peso.
Para conseguir recuperar el peso adecuado, una vida activa y una alimentación sana y adecuada son los mejores métodos.
Ejercicio físico
Otro de los factores a destacar en el proceso de recuperación es el ejercicio físico. Las personas que están siguiendo un tratamiento con quimioterapia experimentan síntomas de agotamiento que a menudo no se alivian con el descanso. Esta fatiga impide a las personas estar activas y desempeñar algunos tipos de actividades.
Para paliar estos síntomas, el ejercicio físico es un excelente método ya que se ha demostrado que los pacientes que llevan a cabo una rutina de ejercicios adaptados a sus necesidades se sienten mejor tanto física como emocionalmente. También se ha comprobado que ayuda a reducir los efectos secundarios del tratamiento, algo de gran importancia para la recuperación.
Salud emocional
Para el paciente, el hecho de ser diagnosticado supone un gran impacto en su vida y puede hacer mella en su estado de ánimo y en sus relaciones con las personas de su entorno.
Los expertos aconsejan que los pacientes busquen apoyo, tanto en el equipo médico con el que están llevando a cabo el tratamiento como con profesionales o grupos que puedan ayudarles desde el punto de vista psicológico o emocional.
En la fase de diagnóstico, se genera un sentimiento de incertidumbre en el paciente que se ve agravado por un pronóstico incierto. En esta fase, es de gran importancia que el paciente recupere el equilibrio emocional.
Con este objetivo, se propone evaluar y promover iniciativas personalizadas para ayudar a los pacientes y familiares a afrontar los cambios que se presentan después del diagnóstico y durante el tratamiento.
Algunas de las iniciativas que se proponen son tanto a nivel personal (trabajo de la autoestima, autoimagen) como en la interactuación con el entorno (apoyo en redes, evitar ruptura brusca con la vida cotidiana). Este seguimiento debe continuar durante todo el proceso evolutivo y todas las fases de la enfermedad, desde el momento del diagnóstico, pasando por el tratamiento, la curación, la recaída y re-tratamiento si se produce, y la fase terminal de la enfermedad en cuidados paliativos.
Durante y tras la finalización del tratamiento, el paciente debe de estar en contacto directo con su médico con el fin de mantener unos controles exhaustivos de su evolución. De este modo se podrá detectar cualquier cambio que pudiera surgir en el estado de salud del paciente, atendiendo tanto a posibles recaídas como a posibles efectos adversos generados por el propio tratamiento.
Con independencia de los hábitos generales aquí descritos, el paciente ha de consultar todas las dudas que tenga con su médico con el fin de adecuar el tratamiento a sus condiciones y requerimientos individuales.
Referencia bibliográfica
- Soledad de Linares Fernández et al. Guía informativa para pacientes hematológicos. Hospital Universitario Virgen de las Nieves.
- Soledad de Linares Fernández et al. Manual de apoyo al paciente Hematológico. Hospital Universitario Virgen de las Nieves.
- Rodríguez-Durán et al. Percepción de la importancia de la alimentación en un grupo de pacientes con cáncer hematológico. Nutr. Hosp. vol.27 no.2 mar./abr. 2012
- Referencia: Dennett, A. M., Peiris, C. L., Shields, N., Prendergast, L. A., & Taylor, N. F. (2016). Moderate-intensity exercise reduces fatigue and improves mobility in cancer survivors: a systematic review and meta-regression. Journal of Physiotherapy.
- Juan Antonio Cruzado et al. Intervención psicológica en un caso de leucemia mieloide aguda con problemas de adaptación a la hospitalización y trastorno psicótico inducido por substancias.
- Arber DA et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. 2016 May 19;127(20):2391-405
- https://www.fcarreras.org/
- https://www.aeal.es/
- https://www.cancer.gov/
- https://www.cancer.org/
- https://bitacoramedica.com/
Macroglobulinemia de Waldenström
¿Qué es la Macroglobulinemia de Waldenström?
La Macroglobulinemia de Waldenström (MW), también conocida como linfoma linfoplasmocítico, es un subtipo de linfoma no Hodgkin de bajo grado de malignidad (de progresión lenta) que afecta los linfocitos (glóbulos blancos).
Su nombre se debe al médico Jan Gösta Waldenström que fue el primero en describirla en el año 1944. Es un cáncer de la sangre muy poco frecuente caracterizado por una proliferación de un grupo (clon) de células de estirpe B (linfocitos, linfoplasmocitos y células plasmáticas) que tienen un comportamiento anormal e invaden la médula ósea y a veces los ganglios linfáticos, bazo y otros órganos. Además son capaces de producir y liberar al suero un único tipo de proteína anormal (monoclonal) en grandes cantidades. Esta proteína es un anticuerpo o inmunoglobulina de tipo M, a la que llamamos IgM monoclonal 1. Por esto, las personas con MW suelen tener la sangre más espesa. Las inmunoglobulinas (Ig), o anticuerpos, son sustancias que circulan por la sangre con el objetivo de defender al individuo, atacando toda materia extraña que entre en el organismo, pero en el caso de la MW no puede cumplir esa finalidad. La MW se puede confundir con algunos linfomas, por lo que para hacer un diagnóstico correcto es necesario descartarlos.

Es importante distinguir la MW de algunos procesos muy similares, que no tienen capacidad maligna y pueden acompañar a una persona durante el resto de su vida sin provocarle ningún problema de salud. Estamos hablando de la denominada “gammapatía monoclonal de significado incierto de tipo GMSI que a veces es muy difícil de distinguir de la MW. Al igual que en la GMSI, hay casos de MW que no provocan síntomas y nunca requieren tratamiento. Para hacer estas distinciones, su médico tendrá que utilizar unos criterios diagnósticos que han sido establecidos por grupos internacionales de trabajo y se pueden consultar en la clasificación actual de la OMS (Organización Mundial de la Salud)
¿Qué causa la MW?
La MW es una enfermedad muy poco frecuente. En España, su incidencia es 3,1 casos por millón de habitantes y año,3 lo que implica que hay unos 150 pacientes nuevos cada año. Esto, unido a la larga supervivencia de los pacientes, hace que haya unos 2000 pacientes a día de hoy en nuestro país. Afecta sobre todo a pacientes mayores (mediana de 71 años) y es un poco más frecuente en los varones (70%).
La causa de la MW se desconoce, pero hay cierta influencia genética o familiar, ya que hay familias con varios miembros diagnosticados de MW. Así, hasta el 19% de los pacientes con MW tienen algún familiar con MW o proceso similar.4 Además, los familiares de primer grado de pacientes con MW tienen un riesgo hasta 20 veces superior a la población general de padecer esta enfermedad,5 si bien sólo supone 6 nuevos casos anuales por cada 10.000 habitantes. También hay datos que avalan su asociación a infecciones (Virus de la Hepatitis C) o autoinmunidad.
Hoy se piensa que la MW se produce mediante un complicado proceso en el que el primer paso es la transformación del linfocito B en una célula inmortal. Esta célula adquiere una mutación muy concreta de la MW que da lugar a una población más o menos numerosa, que aunque es inmortal y acompañará a la persona que la tenga durante toda su vida, no es maligna y no provoca ningún problema de salud7. Si esta población celular tiene unas características genéticas propicias y sufre nuevas anomalías, como la pérdida de parte del cromosoma 6, o la adquisición de otras mutaciones va perdiendo capacidad de control, o lo que es lo mismo, ganando capacidad de multiplicarse y agredir a su entorno, provocando síntomas. Entonces es cuando decimos que ya se ha hecho maligna y se comportará como muchos otros cánceres. Su médico decidirá entonces como abordar la enfermedad y el tratamiento.
¿Cuáles son los signos y síntomas que produce la MW?
La MW suele ser una enfermedad lenta y crónica, pudiendo permanecer estable durante años sin síntomas importantes. Por ello clasificamos a las MW en formas sintomáticas y asintomáticas. Cuando no hay síntomas no es necesario poner tratamiento.
Primer síntoma
El síntoma inicial más frecuente es cansancio progresivo3. A distancia le siguen las hemorragias, las manifestaciones neurológicas y las adenopatías (aumento del tamaño de los ganglios). También puede haber otras manifestaciones menos frecuentes: respiratorias, oculares, insuficiencia renal, síntomas cutáneos, etc. Finalmente, el diagnóstico también puede ser casual en una analítica rutinaria, (como la velocidad de sedimentación globular [VSG] aumentada o hallazgo de proteína monoclonal) en un estudio realizado por otra causa. Ya hemos dicho también que hay casos de Gammapatía Monoclonal de Significado Incierto, de los que una pequeña parte puede transformarse en MW.
Qué síntomas pueden aparecer
Aparte del primer síntoma, la MW puede producir muchos síntomas que los médicos dividen en debidos al crecimiento tumoral y debidos a presencia de la proteína IgM monoclonal.
Manifestaciones por crecimiento tumoral
Poco más de la cuarta parte de los pacientes tienen lo que en medicina se llama un cuadro constitucional, caracterizado por abatimiento, falta de apetito y pérdida de peso3. Dado que el crecimiento de las células malignas se da especialmente en la médula ósea, es frecuente que su función esté reducida, lo que provoca una reducción de las células de la sangre: bajan los glóbulos rojos y la hemoglobina que contienen (anemia), bajan los glóbulos blancos (leucopenia) y bajan las plaquetas (trombopenia). De estos problemas, el más frecuente es la anemia, que la principal causa del cansancio y el motivo más frecuente por el que los pacientes requieren tratamiento.
Alrededor de la quinta parte de los pacientes tiene infiltración de órganos linfoides, es decir ganglios o bazo aumentados de tamaño y que se pueden tocar, y agrandamiento del hígado. No obstante, estos hallazgos suelen ser leves o a lo sumo moderados.
La afectación de otras regiones del cuerpo (infiltración de otros tejidos) es poco frecuente y generalmente causa pocos síntomas3. Se puede afectar la pleura, el pulmón, el riñón o la piel. Otras regiones como hueso, tubo digestivo o tejido nervioso se afectan de forma excepcional.
Manifestaciones clínicas dependientes de la proteína IgM monoclonal
La molécula de IgM es muy grande, se agrega y aumenta la viscosidad de la sangre. Además, por sí sola o con otras proteínas, puede tener comportamientos anormales con síntomas especiales.
La alteración más llamativa por componente monoclonal es el síndrome de hiperviscosidad, que aparece en el 15% de los pacientes con MW.3Generalmente aparece cuando la cantidad de proteína es muy alta, pero no siempre. Los síntomas pueden ser:
- Neurológicos y oculares, donde de menor a mayor gravedad van apareciendo dolor de cabeza, confusión, vértigo, somnolencia, visión borrosa y estupor, pudiendo llegar al coma. Los primeros signos los podemos ver en la retina, al ver dilataciones y estrechamientos de los vasos sanguíneos y otras anomalías. Esto se ve fácilmente con un análisis del fondo del ojo.
- Manifestaciones cardíacas progresivas, desde las más leves como hipertensión arterial no controlable hasta las más graves, como la insuficiencia cardíaca, sobre todo en ancianos.
- Hemorragias, sobre todo en las mucosas.
El 10% de los pacientes tiene una proteína IgM que precipita a bajas temperaturas (crioglobulina). De ellos, la cuarta parte tienen síntomas de crioglobulinemia: fenómeno de Raynaud (cambios dolorosos en el color de los dedos de manos o pies), necrosis de las zonas distales del cuerpo (oreja, nariz, dedos) y manchas por sangrado intracutáneo (púrpura) en extremidades inferiores. Pueden aparecer otras alteraciones en la piel, pero son muy raras. En tal caso consulte a su médico.
Otros posibles efectos
1) Sensibilidad a bajas temperaturas:
Algunos pacientes con MW desarrollan una respuesta dolorosa al frío. Esto sucede porque la sangre es más espesa, como si fuera un gel, debido a los anticuerpos IgM. Cuando ciertas zonas del cuerpo se exponen al frío (por ejemplo los dedos de las manos o de los pies, o la punta de la nariz), pueden sentir mucho dolor. A veces provoca dolor en las articulaciones, problemas renales y puede afectar a la piel.
Esta respuesta se llama “crioglobulinemia” y afecta a un 20% de los pacientes con MW. Incluso entonces solo cerca del 5% experimenta algún síntoma.
2) Espesamiento de la sangre:
La acumulación de IgM hace que la sangre sea más espesa de lo normal, esto interfiere en la circulación porque no puede fluir libremente por los vasos sanguíneos pequeños.
Esto se denomina “síndrome de hiperviscosidad” y entre el 10-30% de las personas con MW lo presenta.
- Si afecta al cerebro puede provocar los mismos problemas que un ictus como debilidad en un lado del cuerpo y dificultad para hablar
- Cuando afecta al corazón puede causar insuficiencia cardiaca congestiva por sobrecarga del corazón
3) Macroglobulinas:
La acumulación de las macroglobulinas puede causar síntomas variados como un sangrado excesivo, problemas de visión y del sistema nervioso.
Diagnóstico de la MW
Como ya hemos dicho antes, hay varios síntomas que harán sospechar a su médico, que algo no está bien y se empezará un estudio en el que irán apareciendo varias anomalías, o directamente, al hacer un análisis aparecerá una anomalía que nos inducirá a pensar en una MW. En ese caso, su médico pedirá varias pruebas complementarias:
- Estudio biológico en la sangre periférica
1.1 Hemograma
Hay anemia en el 50-70% de los enfermos, con un patrón de enfermedad crónica (anomalías por mala utilización del hierro del cuerpo) o infiltrativa (por invasión de la médula ósea). Los glóbulos blancos suelen ser normales (sólo bajan en el 10-20%), pero en la tercera parte de los pacientes puede haber aumento de un tipo de ellos: los linfocitos. Las plaquetas bajan en menos del 20% de los casos.
1.2 Bioquímica
La bioquímica de los pacientes con MW suele ser normal.
1.3 Proteinograma sérico y urinario
Las proteínas totales están aumentadas por la presencia de la proteína IgM monoclonal, y la velocidad está muy aumentada. Al analizar las proteínas se ve la existencia de un pico o banda monoclonal, que al ser identificada por métodos inmunológicos se revela de tipo IgM. Esto constituye una prueba muy útil para hacer bien el diagnóstico, porque aunque la MW se parece al mieloma, el pico monoclonal de éste casi nunca es IgM. La MW también se parece a la leucemia linfocítica crónica, pero en ésta muy pocas veces hay proteína monoclonal. Además, y a diferencia tanto del mieloma como de la leucemia linfocítica crónica, los niveles de las restantes inmunoglobulinas suele ser normales en la MW.3 Por último, también es posible ver restos de la inmunoglobulina monoclonal en la orina, pero sólo en la tercera parte de los pacientes y en muy poca cantidad. Otra determinación importante es la de las cadenas ligeras libres (FLC) en suero, que su médico sabrá manejar correctamente.
- Estudio de Médula Ósea
Para el diagnóstico de MW es necesario realizar una biopsia ósea para demostrar que hay ciertas células que han entrado en la médula ósea (invasión linfoplasmocítica), por lo que esta prueba es obligatoria y será analizada por el patólogo.
En paralelo a la biopsia se realiza un aspirado medular. Además, su hematólogo deberá realizar un estudio con técnicas de última generación con citometría, citogenética y biología molecular de la médula para ver la presencia de otras alteraciones. Debemos destacar: pérdida del brazo largo del cromosoma 6, traslocaciones del cromosoma 14 (14q32) o pérdidas de los cromosomas 13 ó 17, que ayudan mucho a fijar el diagnóstico de la MW9. Sin embargo, hoy día es obligatorio, y cualquier paciente puede exigirlo, un estudio molecular que determine la presencia o no de la mutación MYD88 L265P, muy fácil de obtener y de gran ayuda para el diagnóstico. Posiblemente también tenga valor para predecir la respuesta al tratamiento.
- Estudio de ganglios linfáticos y otros tejidos
Es necesario hacer un TC (escáner) de cuerpo entero para buscar adenopatías y cuantificar organomegalias (órganos agrandados). Se recomienda biopsiar cualquier adenopatía accesible si se sospecha otro tipo de linfoma y así descartarlo.
- Otras exploraciones complementarias
Estudio de fondo de ojo para valorar signos de hiperviscosidad;
Estudio de amiloide (tinción con Rojo Congo) en la biopsia de la médula ósea, grasa peritoneal o mucosa rectal;
Test de Coombs, para descartar la presencia de anemia hemolítica inmune
Determinación de crioaglutininas séricas.
Otras, según clínica: electromiograma, serie ósea, estudio gastro-intestinal, biopsia cutánea,…
Pronóstico de la MW
En un estudio español publicado en 2001, la mediana de la supervivencia fue de 11 años desde el diagnóstico. En 2016 la mediana es probablemente superior a los 15 años. No obstante, varía de unos pacientes a otros. Para intentar predecirla se usan los factores pronósticos.10 Sabemos que la presencia de algunos factores tales como edad avanzada, anemia, trombopenia, albúmina baja, Beta2-microglobulina alta o proteína monoclonal muy alta confieren un peor pronóstico.
No podemos olvidar que más de un tercio de los pacientes, probablemente la mitad, fallecen por causas no relacionadas con la MW. El resto se relacionan con la progresión de la enfermedad (hiperviscosidad, hemorragia, anemia hemolítica, etc.) y los procesos infecciosos secundarios a la inmunosupresión propia de la enfermedad y su tratamiento.
¿Cómo se vive con MW?
El tratamiento de los Macroglobulinemia de Waldeström varía dependiendo de muchos factores. Uno de los principales es la edad biológica, es decir, la edad que corresponde al estado físico y a la salud del paciente.
Múltiples facetas de la vida diaria como la alimentación, nivel de actividad física y estado mental pueden afectar al estado de salud por lo que es muy importante mantener buenos hábitos en dichos ámbitos.
Nutrición
La nutrición es uno de los factores que deben cuidar las personas que están recibiendo un tratamiento. La dieta debe ser equilibrada, el hecho de incluir en ella alimentos indicados por los profesionales, durante el desarrollo del tratamiento y cuando éste finaliza, puede suponer una mejora muy notable haciendo que el paciente se sienta con más energía.
Los efectos secundarios generados en los primeros estadios del tratamiento (vómitos, náuseas, diarreas…) pueden provocar pérdida de apetito y esto conlleva una pérdida de peso involuntaria. Para intentar sobrellevar estos síntomas se aconseja comer porciones pequeñas cada dos o tres horas; estos problemas suelen remitir con el paso del tiempo.
Ejercicio físico
Otro de los factores a destacar en el proceso de recuperación es el ejercicio físico. El ejercicio físico es un excelente método ya que se ha demostrado que los pacientes que llevan a cabo una rutina de ejercicios adaptados a sus necesidades se sienten mejor tanto física como emocionalmente. También se ha comprobado que ayuda a reducir los efectos secundarios del tratamiento, algo de gran importancia para la recuperación.
Salud emocional
Para el paciente, el hecho de ser diagnosticado supone un gran impacto en su vida y puede hacer mella en su estado de ánimo y en sus relaciones con las personas de su entorno.
Los expertos aconsejan que los pacientes busquen apoyo, tanto en el equipo médico con el que están llevando a cabo el tratamiento como con profesionales o grupos que puedan ayudarles desde el punto de visto psicológico o emocional.
En la fase de diagnóstico, se genera un sentimiento de incertidumbre en el paciente que se ve agravado por un pronóstico incierto. En esta fase, es de gran importancia que el paciente recupere el equilibrio emocional.
Con este objetivo, se propone evaluar y promover iniciativas personalizadas para ayudar a los pacientes y familiares a afrontar los cambios que se presentan después del diagnóstico y durante el tratamiento.
Algunas de las iniciativas que se proponen son tanto a nivel personal (trabajo de la autoestima, autoimagen) como en la interactuación con el entorno (apoyo en redes, evitar ruptura brusca con la vida cotidiana).Este seguimiento debe continuar durante todo el proceso evolutivo y todas las fases de la enfermedad, desde el momento del diagnóstico, pasando por el tratamiento, hasta la fase de cuidados paliativos.
Durante y tras la finalización del tratamiento, el paciente debe de estar en contacto directo con su médico con el fin de mantener unos controles exhaustivos de su evolución. De este modo se podrá detectar cualquier cambio que pudiera surgir en el estado de salud del paciente, atendiendo tanto a posibles recaídas como a posibles efectos adversos generados por el propio tratamiento.
Con independencia de los hábitos generales aquí descritos, el paciente ha de consultar todas las dudas que tenga con su médico con el fin de adecuar el tratamiento a sus condiciones y requerimientos individuales.
Con independencia de los hábitos generales aquí descritos, el paciente ha de consultar todas las dudas que tenga con su médico con el fin de adecuar el tratamiento a sus condiciones y requerimientos individuales.
Referencia bibliográfica
Owen RG, Treon SP, Al Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 2003; 30(2): 110-5.
Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of the Haematopoietic nd tymphoid tissues. 4th edition. Lyon: International Agency for Resarch on Cancer; 2008.
Garcia-Sanz R, Montoto S, Torrequebrada A, et al. Waldenstrom macroglobulinaemia: presenting features and outcome in a series with 217 cases. BrJ Haematol 2001; 115(3): 575-82.
Wang H, Zhou Y, Zhang Y, et al. Trend and Geographic Variations in the Incidence of Waldenstrîm’s Macroglobulinemia in the United States over 17 Years from 1988 to 2004. ClinLymphoma Myeloma 2008; 8: (Suppl.):101a.
Kristinsson SY, Bjorkholm M, Goldin LR, McMaster ML, Turesson I, Landgren O. Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia patients: a population-based study in Sweden. Blood 2008; 112(8): 3052-6.
Treon SP, Xu L, Yang G, et al. MYD88 L265P Somatic Mutation in Waldenstrîm’s Macroglobulinemia. New England Journal of Medicine 2012; 367(9): 826-33.
Jimenez C, Sebastian E, Chillon MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom’s macroglobulinemia. Leukemia 2013; 27(8): 1722-8.
Paiva B, Montes MC, Garcia-Sanz R, et al. Multiparameter flow cytometry for the identification of the Waldenstrom’s clone in IgM-MGUS and Waldenstrom’s Macroglobulinemia: new criteria for differential diagnosis and risk stratification. Leukemia 2014; 28(1): 166-73.
Braggio E, Philipsborn C, Novak A, Hodge L, Ansell S, Fonseca R. Molecular pathogenesis of Waldenstrîm macroglobulinemia. Haematologica 2012; 97(9): 1281-90.
Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood 2009; 113(18): 4163-70.
Kyle RA, Treon SP, Alexanian R, et al. Prognostic markers and criteria to initiate therapy in Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. SeminOncol 2003; 30(2): 116-20.
Dimopoulos MA, Kastritis E, Owen RG, et al. Treatment recommendations for patients with Waldenstrom macroglobulinemia (WM) and related disorders: IWWM-7 consensus. Blood 2014; 124(9): 1404-11.
Treon SP. How I treat Waldenstrom macroglobulinemia. Blood 2015; 126(6): 721-32.
En Cómo se vive con la patología: Soledad de Linares Fernández et al. Guía informativa para pacientes hematológicos. Hospital Universitario Virgen de las Nieves. // Soledad de Linares Fernández et al. Manual de apoyo al paciente Hematológico. Hospital Universitario Virgen de las Nieves. // D. Rodríguez-Durán et al. Percepción de la importancia de la alimentación en un grupo de pacientes con cáncer hematológico. Nutr. Hosp. vol.27 no.2 mar./abr. 2012 // Dennett, A. M., Peiris, C. L., Shields, N., Prendergast, L. A., & Taylor, N. F. (2016). Moderate-intensity exercise reduces fatigue and improves mobility in cancer survivors: a systematic review and meta-regression. Journal of Physiotherapy. // Juan Antonio Cruzado et al. Intervención psicológica en un caso de leucemia mieloide aguda con problemas de adaptación a la hospitalización y trastorno psicótico inducido por substancias.